To center or not to center?¶
Summary¶
Pytim provides different ways to center the interface in the simulation box, and how to compute profiles.
This short tutorial shows the different behaviors.
Example: pdb output for a planar interface¶
Initial file
The waterbox example file WATER_GRO consists of 4000 water molecules, with all
atoms in the unit cell, and the liquid phase crossing the periodic boundary conditions:
If you want to save a local copy of the file, you just need to use this code fragment:
import MDAnalysis as mda
import pytim
u = mda.Universe(pytim.datafiles.WATER_GRO)
u.atoms.write('centering1.pdb')
When initializing/computing the interface, Pytim internally centers the interface in different ways, depending whether ITIM or GITIM is used. When the configuration with the surface layers information is written to file using writepdb(), the system can be shifted so that the liquid phase is placed at different positions.
Default, no centering
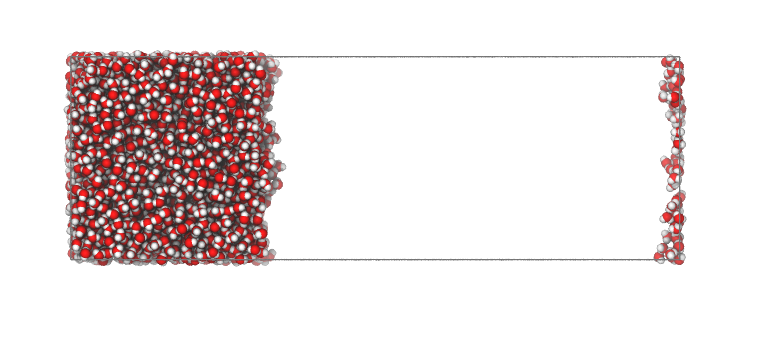
If the (default) option centered='no' is used, then all atomic positions are kept the same as in the input file.
This is like the initial case, however with information on the surface molecules added to the pdb.
interface = pytim.ITIM(u)
interface.writepdb('centering2.pdb',centered='no')
Centering at the origin
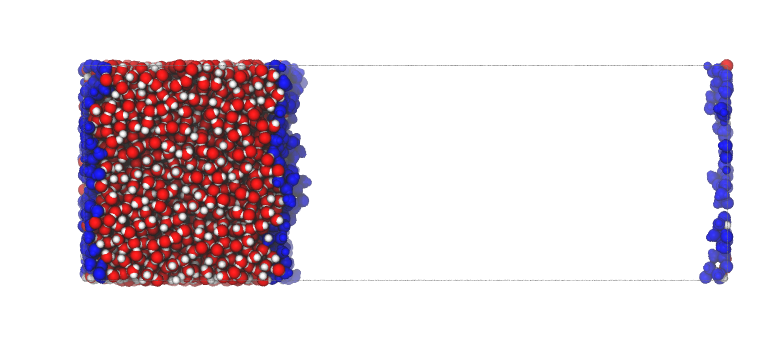
The system can be shifted so that the liquid phase is placed across the origin of the normal axis, using the option centered=origin.
This comes handy to quickly discriminate between upper and lower surface atoms, to spot immediately the middle of the liquid phase, or to verify that the automatic centering method behaved as expected.
interface = pytim.ITIM(u)
interface.writepdb('centering3.pdb',centered='origin')
Centering in the middle
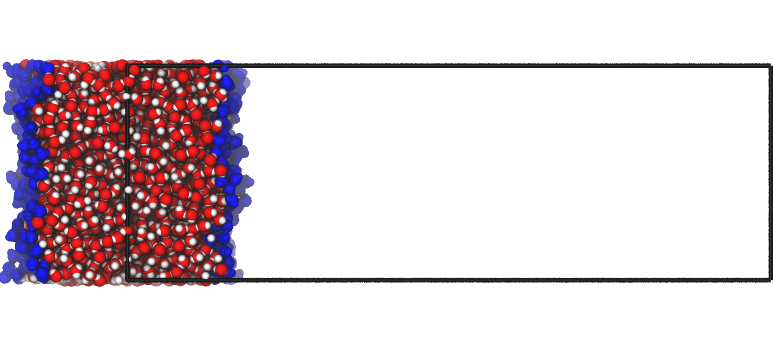
If the option centered='middle' is passed to writepdb(), instead, then the liquid part of the system is placed in the middle of the box along the surface normal:
interface.writepdb('centering4.pdb',centered='middle')
